Bacterial typing methods
In this exercise we will be focusing on reference based genome analysis. This is, by far, not the only analysis possible and is only one of many different systems for typing and characterising bacterial genomes from sequence data. For an overview of typing methods see Authhors.
Bactopia for reference based genome analysis
Bactopia provides a snippy workflow that is aimed at reference based genome
analysis using snippy,
gubbins,
snp-dists and IQ-Tree.
Choosing a reference genome
The choice of a reference genome depends a lot on the biology of the organism being analysed. Some bacteria are highly clonal. A good example is M. tuberculosis, where the H37Rv genome, or the inferred ancestral genome are typically used as reference genomes.
In other species such as E. coli, high diversity in the species and widespread horizontal gene transfer and recombination prevent the use of a single reference genome. In the case of an outbreak of disease an ‘outbreak genome’ is often chosen or produced against which all outbreak samples are compared. Tools such as referenceseeker can be used to find a closely related reference genome, but such choices should be made with caution to avoid selecting an innapropriate or poor quality reference.
As 7th pandemic cholera is caused by the rapid expansion and worldwide distribution of a successful strain of V. cholerae, the N16961 strain, collected in Bangladesh in 1975. The sequence of this strain was first published in 2000 and a recent publication by Matthey et al corrected an inversion in the initial assembly using PacBio long reads, leading to CP028827 and CP028828, the sequences of the large and small chromosomes of the bacterium. These are commonly used as reference sequences when analysing 7th pandemic El Tor (7PET) cholera samples.
For the purposes of our analysis we will follow the approach used by the authors of the Mavian et al paper and use NC_016445 and NC_016446, the sequences of the two chromosomes of the 2010EL-1786 strain as a reference. This isolate was collected in Haiti, early in the outbreak.
For reference based analysis we need a single sequence as the reference, and
for snippy to annotate the VCF we need that reference to be in Genbank format.
Thus the two sequences were downloaded from Genbank (using the ‘Send to’
menu on the NCBI website and selecting ‘File’ output and ‘Genbank (full)’ as
the format). The two chromosomes were merged using the merge-gbk-records
tool (installed from Bioconda using conda) with the command:
merge-gbk-records -l0 NC_016445.genbank NC_016446.genbank >combined.genbank
Setting up your Bactopia run
The previously used bactopia_common.sh will be re-used to store the basic
settings for Bactopia. The snippy workflow will be run using:
#!/bin/bash
BACTOPIA_DIR=cleaned_reads
. bactopia_common.sh
$BACTOPIA --wf snippy --reference combined.genbank --bactopia $BACTOPIA_DIR --run_name NC_016445_NC_0164456 --outdir bactopia_snippy
This will take the previously prepared reads from the cleaned_reads directory and align them against the combined.genbank
reference, with output being written to the bactopia_snippy directory. Output is organised in directories named like:
bactopia_snippy/SAMPLE1/tools/snippy/NC_016445_NC_0164456/
bactopia_snippy/SAMPLE2/tools/snippy/NC_016445_NC_0164456/
bactopia_snippy/bactopia-runs/snippy-YYYYMMDD-HHMMSS/
If the workflow is run multiple times, multiple bactopia-runs directories will be created. Keep track of the times of the
runs to ensure that you select the correct output directory. In the rest of the notes the appropriate directory in
bactopia-runs will simply be referred to as the bactopia-runs directory.
The --run_name parameter is necessary to give a meaningful output name to the directory in which the snippy results are
stored. If no --run_name parameter is used, the results will be in a tools/snippy/snippy folder for each sample.
The snippy tool combines the steps of read mapping (using bwa mem), variant calling (using freebayes) and variant
annotation (using snpeff) together with various post-processing and filtering steps. The files that snippy creates are
largely identical to the ones described on the snippy github page. Some key outputs are:
SAMPLE.consensus.fa.gz- the reference genome with all variants insertedSAMPLE.consensus.subs.fa.gz- there reference genome with only single nucleotide variants insertedSAMPLE.bamandSAMPLE.bam.bai- the aligned reads in BAM format and BAM indexSAMPLE.SRR8364253.annotated.vcf.gz- filtered variants annotated usingsnpeff
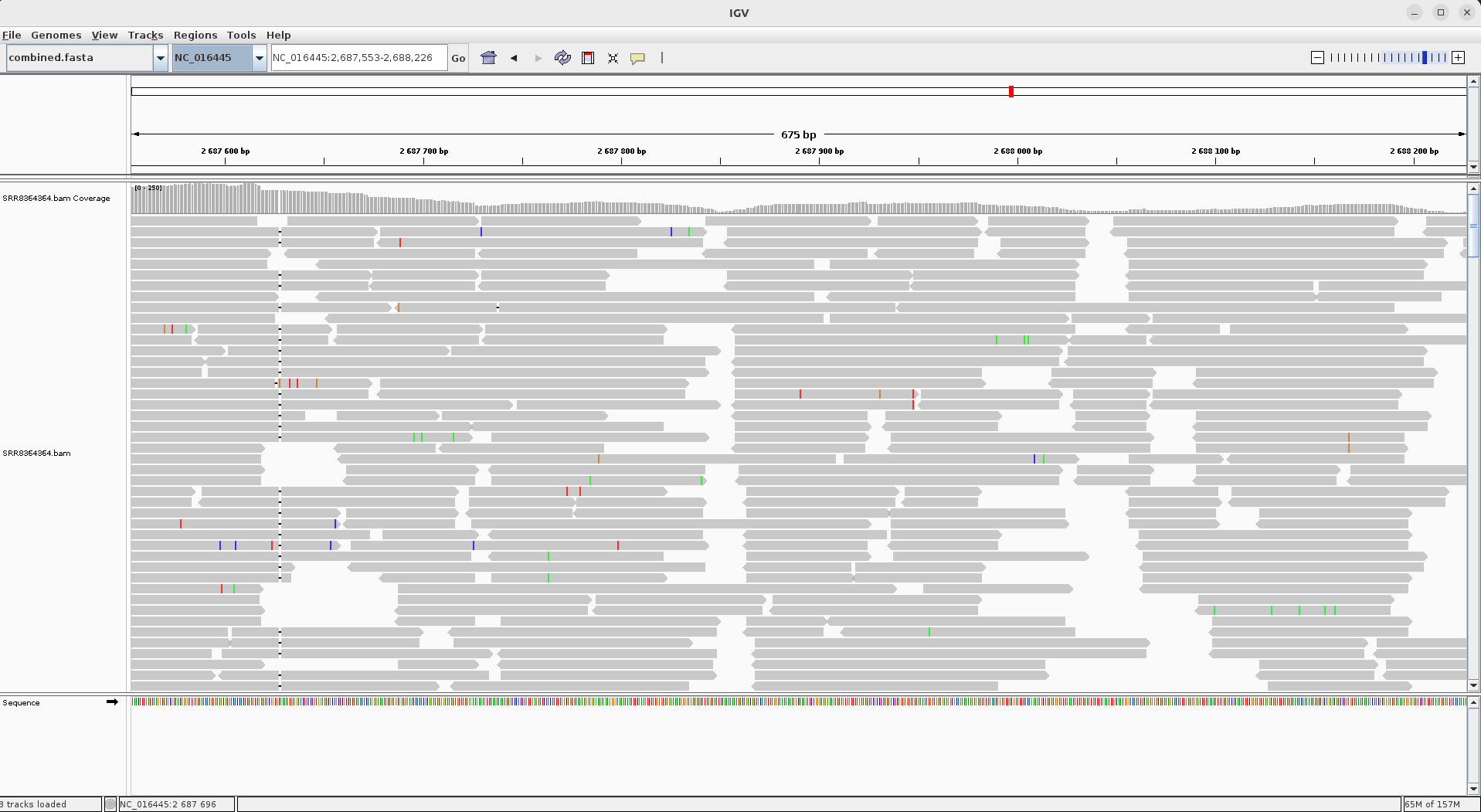
Browsing the mapped reads using IGV
The BAM file can be visualised with a BAM viewer like IGV but note that it is in the coordinate scheme of the combined
references (combined.genbank) and that you need to create a FASTA file of the reference genome using e.g. any2fasta (which can be
installed using conda):
any2fasta combined.genbank >combined.fasta
This genome can then be loaded using IGV’s Genomes -> Load Genome from File menu item. In the screenshot below (of sample SRR8364364) the area
‘NC_016445:2,687,553-2,688,226’ is show. The one base pair deletion that leads to the O1 Igawa serotype is visible at the left of the picture.
If you want to see the positions of genes and other features in IGV, you can create a GFF3 file from the combined.genbank using the seqret
tool from the emboss package (conda create -n emboss emboss):
seqret 'genbank::combined.genbank' -outseq 'gff3::combined.gff3' -feature
Load this GFF3 file using the Load File menu item of IGV and right-click on the track and set it to Expanded.`
In the next section we will examine the outputs in the bactopia-runs directory and discuss integrating the bactopia outputs with
other tools.