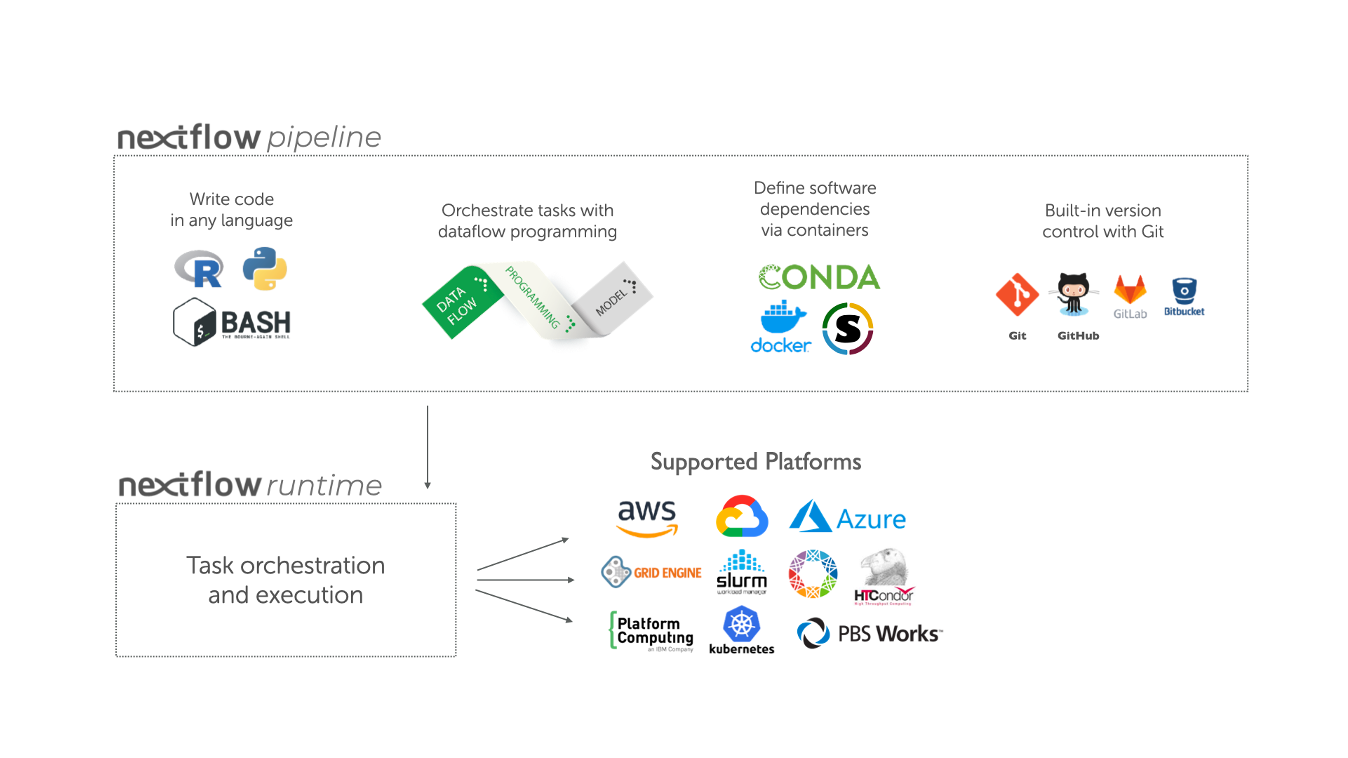
Processes, channels and workflows
Nextflow workflows have three main parts; processes, channels, and workflows.
-
Processes describe a task to be run. A process script can be written in any scripting language that can be executed by the Linux platform (Bash, Perl, Ruby, Python, etc.). Processes spawn a task for each complete input set. Each task is executed independently and cannot interact with another task. The only way data can be passed between process tasks is via asynchronous queues, called channels. Processes define inputs and outputs for a task.
-
Channels are then used to manipulate the flow of data from one process to the next.
-
The interaction between processes, and ultimately the pipeline execution flow itself, is then explicitly defined in a workflow section.
Processes and channels
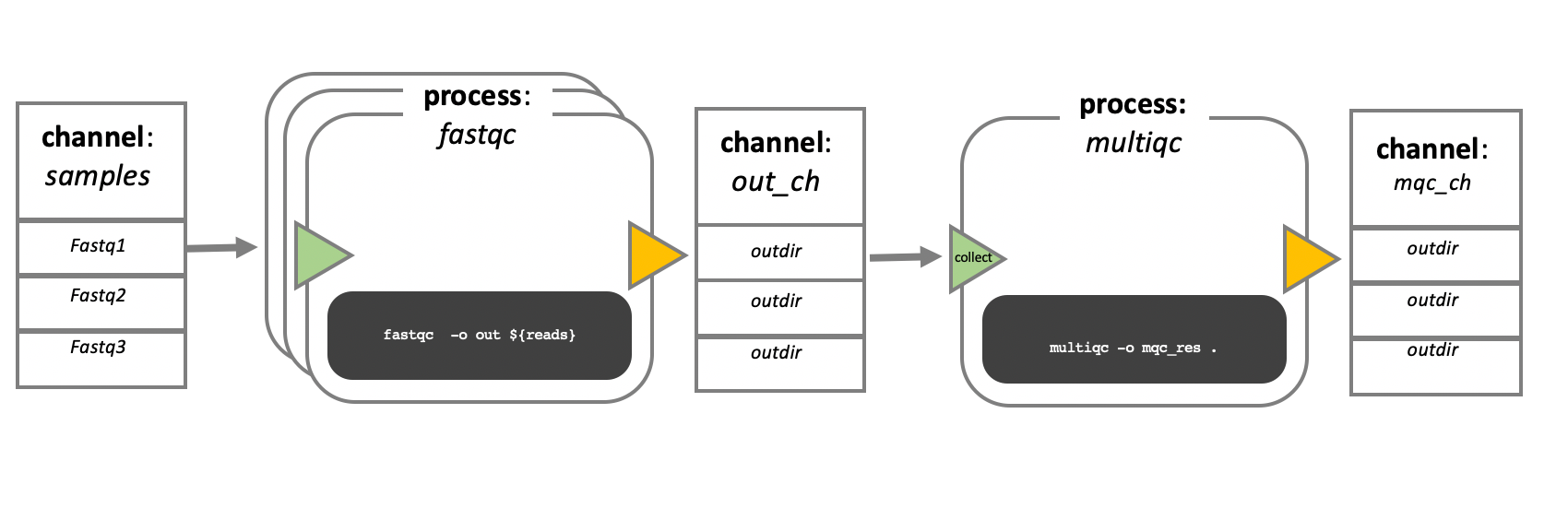
In the example below, we have a channel containing three elements, e.g., 3 data files. We have a process that takes the channel as input. Since the channel has three elements, three independent instances (tasks) of that process are run in parallel. Each task generates an output, which is passed to another channel and used as input for the next process.
Workflow execution
While a process defines what command or script must be executed, the executor determines how that script is run in the target system.
If not otherwise specified, processes are executed on the local computer. The local executor is very useful for pipeline development, testing, and small-scale workflows, but for large scale computational pipelines, a High-Performance Cluster (HPC) or Cloud platform is often required.
Process execution block
nextflow.enable.dsl=2
process <NAME> {
[ directives ]
input:
< process inputs >
output:
< process outputs >
when:
< condition >
[script|shell|exec]:
< user script to be executed >
}
Example of running a shell command
#!/usr/bin/env nextflow
nextflow.enable.dsl = 2
process INDEX {
cpus 4
input:
path transcriptome
output:
path ‘salmon_index’
script:
""”
salmon index –threads $task.cpus –t $transcriptome –i salmon_index
"""
}
Example of running a python script
//process_python.nf
nextflow.enable.dsl=2
process PYSTUFF {
script:
"""
#!/usr/bin/env python
import gzip
reads = 0
bases = 0
with gzip.open('${projectDir}/data/yeast/reads/ref1_1.fq.gz', 'rb') as read:
for id in read:
seq = next(read)
reads += 1
bases += len(seq.strip())
next(read)
next(read)
print("reads", reads)
print("bases", bases)
"""
}
workflow {
PYSTUFF()
}
Running a nextflow script
nextflow run <script_name> <options/parameters>
nextflow run index_transcriptome.nf
Summary